Νόσος Fabry (Fabry Disease)
Συγγραφέας iator Γράφτηκε στις ,

Η νόσος Fabry ανήκει στα λυσοσωμικά αθροιστικά νοσήματα (Lysosomal Storage Disorders, LSD’s) που περιλαμβάνουν πάνω από 50 σπάνια, γενετικά κληρονομούμενα μεταβολικά νοσήματα με συνολική επίπτωση περίπου 1:7700 γεννήσεις στο γενικό πληθυσμό. Στα λυσοσωμικά αθροιστικά νοσήματα συμβαίνουν διαταραχές στη λειτουργία ενός λυσοσωμικού ενζύμου που έχουν σαν αποτέλεσμα τη συσσώρευση ουσιών (υποστρωμάτων-στόχων) με επακόλουθο τη χρόνια και προοδευτική φθορά των κυττάρων, ιστών και οργάνων που επηρεάζονται.
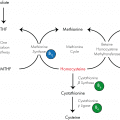
Η νόσος του Fabry, μια από τις τρεις φυλοσύνδετες LSDs, αφορά τη διαταραχή του μεταβολισμού των γλυκοσφιγγολιπιδίων. Οφείλεται στη μερική ή ολική ανεπάρκεια του λυσοσωμικού ενζύμου α-γαλακτοσιδάση Α (a-GAL A) η οποία οδηγεί σε σταδιακή συσσώρευση ουδέτερων αδιάσπαστων γλυκοσφιγγολιπιδίων και ιδιαίτερα του σφαιροτριαζυλοκεραμιδίου Gb-3 στους ιστούς και στο πλάσμα των ασθενών. Τα γλυκοσφιγγολιπίδια εναποτίθενται σε όλη την επιφάνεια του σώματος και κυρίως στα λυσοσώματα των ενδοθηλιακών, επιθηλιακών και λείων μυϊκών κυττάρων του τοιχώματος των αιμοφόρων αγγείων. Οι εναποθέσεις λιπιδίων παρατηρούνται επίσης στα επιθηλιακά κύτταρα του κερατοειδούς, στο σύνολο των κυττάρων των νεφρικών σπειραμάτων και σωληναρίων, στις καρδιακές μυϊκές ίνες και στα γαγγλιοκύτταρα του αυτόνομου νευρικού συστήματος. Η συσσώρευση του κεραμιδίου οδηγεί αρχικά σε δυσλειτουργία των βασικών μεταβολικών διαδικασιών μέσα στο κύτταρο, φτάνοντας έτσι σε κυτταρικό θάνατο που οδηγεί τελικά σε προοδευτική δυσλειτουργία ζωτικών οργάνων.
Κλινική εικόνα
Η νόσος Fabry είναι πολυσυστηματική και απειλητική για τη ζωή διαταραχή του μεταβολισμού των γλυκοσφιγγολιπιδίων, η οποία ως Χ-φυλοσύνδετη νόσος προσβάλλει κυρίως άνδρες ημιζυγώτες αλλά και για τις γυναίκες φορείς.
Τα πρώτα συμπτώματα της νόσου εμφανίζονται συνήθως κατά τη παιδική ή πρώιμη εφηβική ηλικία. Παρότι οι κλινικές εκδηλώσεις της νόσου είναι ιδιαίτερα χαρακτηριστικές, στις περισσότερες περιπτώσεις η διάγνωση καθυστερεί σημαντικά. Ενδεικτικά σε μια πρόσφατη μελέτη που αφορούσε την καταγραφή κλινικών συμπτωμάτων σε 1765 ασθενείς , σημειώνεται ότι η μέση ηλικία έναρξης των συμπτωμάτων ήταν το 9ο έτος στους άντρες και το 13ο έτος στις γυναίκες, ενώ η ηλικία διάγνωσης ήταν κατά το 23ο και το 32ο έτος αντίστοιχα. Οι πλέον συχνές εκδηλώσεις της νόσου στους άντρες ήταν ο διάχυτος πόνος (62%), οι δερματικές αλλοιώσεις (31%), οι γαστρεντερικές διαταραχές (19%), νεφρική προσβολή (17%) και προβλήματα των οφθαλμών (11%). Στις γυναίκες, ο πόνος εμφανιζόταν σε ποσοστό 41%, οι γαστρεντερικές διαταραχές σε ποσοστό 13%, ενώ οι δερματικές αλλοιώσεις και οι διαταραχές των οφθαλμών σε ποσοστό 12%.
Τα συμπτώματα της νόσου παρουσιάζουν χρονική εξέλιξη δεδομένου ότι η συσσώρευση των αδιάσπαστων γλυκολιπιδίων επηρεάζει σε διαφορετικό βαθμό τα διάφορα όργανα Έτσι η εξέλιξη της νόσου μπορεί να χωριστεί σε τρεις περιόδους.
Η αρχική περίοδος κατά τη παιδική-εφηβική ηλικία χαρακτηρίζεται από διαλείποντα επεισόδια ακροπαραισθησιών, υποϊδρωσίας, πυρετού και κόπωσης ενώ εμφανίζονται και τα χαρακτηριστικάαγγειοκερατώματα . Αυτά είναι σκουρόχρωμες ερυθρές έως κυανομέλανες ομάδες αγγειεκτασιών, που καταλαμβάνουν κυρίως τη περιοχή των γεννητικών οργάνων, των γλουτών, καθώς και τη κοιλιακή χώρα και τα άκρα.
Στη δεύτερη περίοδο, μέχρι την ηλικία των 30 ετών, παρατηρείται εξέλιξη των δερματικών αλλοιώσεων σε μέγεθος και αριθμό, ενώ εμφανίζεται και λευκωματουρία σαν πρώτη ένδειξη νεφρικής συμμετοχής. Οι διαταραχές των οφθαλμών, που αφορούν θολερότητα του κερατοειδούς- η οποία με τη πάροδο του χρόνου αποκτά μια χαρακτηριστική σύσπαση (cornea verticillata)- και του αμφιβληστροειδούς, είναι ήδη εμφανείς όμως συχνά παραμένουν αδιάγνωστες επειδή δεν προκαλούν διαταραχές στην όραση.
Η τρίτη περίοδος μεταξύ του 30ου και 50ου έτους χαρακτηρίζεται από την εμφάνιση συμπτωμάτων που υποδηλώνουν την προσβολή πολλαπλών οργάνων και ιστών. Μεταξύ των κλινικών εκδηλώσεων αναφέρονται οι σοβαρού βαθμού καρδιαγγειακές επιπλοκές (αρρυθμίες, στηθάγχη, υπερτροφία της αριστερής κοιλίας, συμφορητική καρδιακή ανεπάρκεια), οι εκδηλώσεις από το νευρικό σύστημα με έντονες ακροπαραισθησίες και κρίσεις γενικευμένου άληγους και η συμμετοχή των νεφρών που καταλήγει σε τελικού σταδίου χρόνια νεφρική ανεπάρκεια (ΤΣΧΝΑ)
Γενετική Διαταραχή – Γονίδιο α-γαλακτοσιδάσης Α
Η συχνότητα εμφάνισης της νόσου υπολογίζεται σε ένα περιστατικό στις 117.000 γεννήσεις ή ένας στους 40.000 – 60.000 άνδρες . Το υπεύθυνο-για τη σύνθεση του λυσοσωμικού ενζύμου α-γαλακτοσιδάση Α – γονίδιο ονομάζεται GLΑ και εντοπίζεται στο μακρύ σκέλος του χρωμοσώματος Χ, στη θέση 22.1 (Xq22.1). Το γονίδιο έχει μέγεθος 12kb περίπου και περιέχει 7 εξόνια τα οποία συνδέονται με εκτεταμένες ρυθμιστικές αλληλουχίες (εσώνια) (5′) καθώς και ανάλογες πλευρικές (3′).
Το εξόνιο 1 περιέχει όλη την 5′ μη-μεταφραζόμενη περιοχή, ενώ κωδικοποιεί το πεπτίδιο σηματοδότησης και τη σειρά των πρώτων 33 αμινοξέων του πρόδρομου ενζύμου. Το γονίδιο περιέχει σημαντικό αριθμό όμοιων επαναλαμβανόμενων νουκλεοτιδικών αλληλουχιών (12 Alu στοιχεία) τα οποία είναι κατανεμημένα στα ιντρόνια και στις πλευρικές περιοχές (3′). Τα στοιχεία αυτά καταλαμβάνουν το 30% του συνολικού μεγέθους του γονιδίου και το καθιστούν ένα από τα πλέον πλούσια ανθρώπινα γονίδια σε περιεκτικότητα αλληλουχιών Alu (~ 1 αλληλουχία Alu / kb). Το πρωτεινικό παράγωγο του γονιδίου, το ένζυμο της α-γαλακτοσιδάσης Α, είναι μια ομοδιμερής γλυκοπρωτεϊνη μοριακού βάρους περίπου 101kDa, η οποία στη τελική της μορφή περιέχει 398 αμινοξέα.
Σε ένα ποσοστό ασθενών οι υπεύθυνες μεταλλάξεις είναι μικρού ή μεγαλύτερου βαθμού γονιδιακές αναδιατάξεις. Τέλος, ατέλειες κατά την επεξεργασία του RNA και μεμονωμένες σύνθετες γονιδιακές βλάβες συμπληρώνουν το σύνολο των μεταλλάξεων του γονιδίου GLA οι οποίες είναι υπεύθυνες για την εμφάνιση της νόσου Fabry.
Τύπος Κληρονομικότητας
Οι ημιζυγώτες άντρες νοσούν στο σύνολό τους και ενώ δεν κληρονομούν τη νόσο στους γιους τους, οι κόρες τους είναι υποχρεωτικά φορείς εφόσον κληρονομούν από το πατέρα τους το Χ χρωμόσωμα με το ελαττωματικό γονίδιο. Με τη σειρά τους οι ετερόζυγες κόρες του πάσχοντα πατέρα, εμφανίζουν 50% πιθανότητα να μεταβιβάσουν τη νόσο στα παιδιά τους
Για πολλά χρόνια θεωρείτο ότι η FD είναι φυλοσύνδετη υπολειπόμενη νόσος, όμως το ψηλό ποσοστό της νόσου στις γυναίκες ετεροζυγώτες υποδεικνύει ότι πρόκειται για νόσο με επικρατητικό χαρακτήρα. Συνήθως τα συμπτώματα στις γυναίκες είναι λιγότερο σοβαρά σε σχέση με τους άντρες ημιζυγώτες αλλά δεν είναι σπάνια η πλήρης εμφάνιση της κλασικής κλινικής έκφρασης της νόσου με σημαντική καρδιακή και νεφρική ανεπάρκεια . Η έντονη κλινική ετερογένεια στις γυναίκες φορείς της νόσου αν και εξαρτάται από το τύπο της εμφανιζόμενης μετάλλαξης, κυρίως οφείλεται στο φαινόμενο της αδρανοποίησης του Χ χρωμοσώματος.
Διάγνωση
Η δυνατότητα για τη τεχνητή παραγωγή ενζύμου σε κυτταρικές σειρές και η έναρξη θεραπείας ενζυμικής υποκατάστασης σε ασθενείς με νόσο Fabry έκανε επιτακτική την ανάγκη άμεσης και ακριβούς διάγνωσης της νόσου προκειμένου οι ασθενείς να υποβληθούν σε θεραπεία προτού οδηγηθούν σε μη αναστρέψιμη οργανική βλάβη.
Η διάγνωση της νόσου βασίζεται κυρίως στον ενδελεχή έλεγχο του οικογενειακού και ατομικού ιστορικού, αλλά και στην ορθή εκτίμηση των συμπτωμάτων που εμφανίζουν οι ασθενείς. Σε παιδιά που παρατηρούνται αγγειοκερατώματα, ακροπαραισθησίες συνοδευόμενες από πυρετό που δεν υποχωρεί, δυσανεξία στο κρύο ή στη ζέστη αλλά και ανεξήγητες γαστρεντερικές διαταραχές ή/και ήπια πρωτεϊνουρία θα πρέπει να τίθεται υποψία της νόσου. Σε ενήλικα άτομα, η παρουσία ανεξήγητης πρωτεϊνουρίας, η έκπτωση της νεφρικής λειτουργίας καθώς επίσης και η εμφάνιση αρρυθμιών, υπερτροφίας αριστερής κοιλίας, αγγειακών εγκεφαλικών επεισοδίων σε νεαρή ηλικία και θολερότητας του κερατοειδούς είναι ενδεικτικές της πιθανής ύπαρξης νόσου Fabry. Η εργαστηριακή επιβεβαίωση της νόσου πραγματοποιείται με μέτρηση της δραστικότητας του ενζύμου της α-γαλακτοσιδάσης στο πλάσμα ή στα λευκά αιμοσφαίρια του αίματος των ασθενών. Οι πάσχοντες από τη κλασική μορφή της νόσου εμφανίζουν συνήθως μηδενική δραστικότητα του ενζύμου ενώ στις άτυπες παραλλαγές είναι δυνατό να βρεθεί μειωμένη δραστικότητα του ενζύμου της τάξης του 5-35% του φυσιολογικού. Αντίθετα για τη επιβεβαίωση της νόσου στις γυναίκες δεν επαρκεί ο βιοχημικός προσδιορισμός των επιπέδων της α-γαλακτοσιδάσης Α, διότι πολύ συχνά η δραστικότητα του ενζύμου στο πλάσμα είναι φυσιολογική. Αναφορικά με τις γυναίκες, εκτός από την εμφάνιση των απαραίτητων κριτηρίων της νόσου πρέπει να γίνεται ανίχνευση της υπεύθυνης μετάλλαξης προκειμένου να τεθεί η διάγνωση .
Προγεννητικός έλεγχος
Είναι εφικτός και ασφαλής ο προγεννητικός έλεγχος της νόσου. Πραγματοποιείται είτε με μέτρηση της δραστικότητας του ενζύμου είτε με την ανίχνευση της ίδιας της μετάλλαξης ήδη από το πρώτο τρίμηνο κύησης σε δείγματα χοριακών λαχνών (βιοψία τροφοβλάστης) ή το δεύτερο τρίμηνο σε δείγματα αμνιακών κυττάρων (αμνιοκέντηση).
Θεραπεία
Η ταυτοποίηση του υπεύθυνου γονιδίου για τη σύνθεση του ενζύμου της α-γαλακτοσιδάσης και η εξέλιξη των τεχνικών του ανασυνδυασμένου DNA επέτρεψαν την εργαστηριακή παραγωγή ικανών ποσοτήτων ενζύμου που έδωσαν το έναυσμα για τη μελέτη της αποτελεσματικότητας της εξωγενούς χορήγησης του ενζύμου ως μορφή θεραπευτικής αντιμετώπισης της νόσου.
Από το 2001 και μέχρι στιγμής υπάρχουν διαθέσιμα δύο φαρμακευτικά σκευάσματα που χρησιμοποιουνται ως θεραπεία ενζυμικής υποκατάστασης. Πρόκειται για ανασυνδυασμένες μορφές του ενζύμου της α-γαλακτοσιδάσης Α (Enzyme Replacement Therapy, ERT) που προκύπτουν είτε απο ανθρώπινες κυτταρικές σειρές, είτε από κύτταρα κινεζικού χοιριδιου και η θεραπεία περιλαμβάνει την ενδοφλέβια χορήγηση του ενζύμου κάθε δυο εβδομάδες.
Η θεραπεία ενζυμικής υποκατάστασης έχει εγκριθεί από το σύνολο των συστημάτων υγείας σε όλο τον κόσμο. Ήδη από τις πρώτες κλινικές μελέτες έχει αποδειχθεί η αποτελεσματικότητα και η ασφάλεια της εξωγενούς χορήγησης της ανασυνδυασμένης α-γαλακτοσιδάσης Α με τη μείωση των επιπέδων του αδιάσπαστου Gb3 στο πλάσμα και στους ιστούς των ασθενών που έχουν μελετηθεί.
ΤΙ ΠΡΟΣΦΕΡΕΙ ΤΟ ΕΡΓΑΣΤΗΡΙΟ ΙΑΤΡΙΚΗΣ ΓΕΝΕΤΙΚΗΣ ΤΟΥ ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ ΣΤΗ ΜΟΡΙΑΚΗ ΜΕΛΕΤΗ ΤΗΣ ΝΟΣΟΥ FABRY
Το Εργαστήριο Iατρικής Γενετικής του Πανεπιστημίου Αθηνών αποτελεί σήμερα κέντρο αναφοράς για τον ελληνικό χώρο για τη μοριακή ανάλυση του γονιδίουGLA σε ασθενείς και οικογένειες με νόσο Fabry. Οι εργαστηριακές εξετάσεις περιλαμβάνουν:
- Ανίχνευση μεγάλων ελλειμμάτων μετά από πολλαπλασιασμό των 7 εξωνίων του γονιδίου GLA με εφαρμογή της αλυσιδωτής αντίδρασης πολυμεράσης (Polymerase Chain Reaction , PCR).
- Εφαρμογή τεχνικής ECMA (Εnzymatic Cleavage Missmatch Anallysis) για την αποκάλυψη πιθανών αλλοιώσεων κατά μήκους του γονιδίου
- Ανάλυση της πρωτοταγούς δομής του DNA (sequencing) για την ταυτοποίηση και τον χαρακτηρισμό των μεταλλάξεων
- Ανίχνευση φορέων στις οικογένειες των ασθενών άμεσα όταν είναι γνωστή η μετάλλαξη στον ασθενή
- Προγεννητικός έλεγχος με μελέτη του DNA του εμβρύου που απομονώνεται από κύτταρα τροφοβλάστης ή κύτταρα αμνιακού υγρού
http://www.iatrikigenetiki.med.uoa.gr/