Με την Ημερίδα σχετικά με τα Σπάνια Νοσήματα του Αναπνευστικού και τα Σπάνια Γενετικά Νοσήματα, ολοκληρώθηκε ο κύκλος Επιστημονικών Συναντήσεων του α΄ εξαμήνου/2013 της Επιστημονικής Εταιρείας Σπανίων Παθήσεων & Ορφανών Φαρμάκων (Ε.Ε.Σ.Π.Ο.Φ.), το Σάββατο 15 Ιουνίου 2013, στην Αθήνα, στην Αίγλη Ζαππείου.
Οι άντρες των οποίων οι σύντροφοι πάσχουν από μία κολπική λοίμωξη που ονομάζεται βακτηριακή κολπίτιδα θα πρέπει να κάνουν κι εκείνοι θεραπεία, σύμφωνα [...]
Ψηφιακοί κίνδυνοι και Σεξουαλική Αυτοεκτίμηση Σε έναν κόσμο στον οποίο η ψηφιακή επανάσταση έχει καταστήσει την πληροφορία, αλλά και κάθε διαστρέβλωση [...]
Το Πάσχα πλησιάζει , αν και διαφορετικό φέτος ωστόσο τα έθιμα από τις περισσότερες οικογένειες θα τηρηθούν για να μας δώσουν την ελπίδα πως σύντομα η [...]
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Η νόσος της Μεσογείου
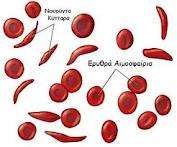
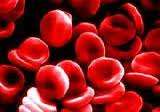
Η μεσογειακή αναιμία ή θαλασσαιμία είναι ένα κληρονομικό νόσημα του αίματος με μεγάλη συχνότητα στη χώρα μας, αλλά και σε άλλες μεσογειακές χώρες. Oι ασθενείς δεν μπορούν να συνθέσουν τη φυσιολογική μορφή της αιμοσφαιρίνης (κύριο συστατικό των ερυθρών αιμοσφαιρίων), με αποτέλεσμα να σχηματίζονται ανώμαλα ερυθρά κύτταρα με μικρή διάρκεια ζωής. Η συχνότητα των φορέων για την Α και Β μεσογειακή αναιμία στην Ελλάδα είναι 8%. Τι συμβαίνει: Η νόσος χαρακτηρίζεται από βαριά αναιμία που εκδηλώνεται από τη βρεφική ηλικία, αλλά οι ασθενείς αντιμετωπίζουν και άλλα προβλήματα, όπως χρόνια κόπωση, δυσκολία στην αναπνοή, οστικές αλλοιώσεις,
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Η νόσος των λευκών
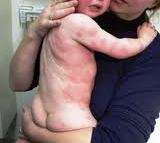
Πρόκειται για την κυστική ίνωση, ένα νόσημα που εμφανίζεται αρκετά συχνά στη λευκή φυλή. Χαρακτηρίζεται από δυσλειτουργία των εξωκρινών αδένων και επηρεάζει το αναπνευστικό και το πεπτικό σύστημα, καθώς και τους ιδρωτοποιούς αδένες. Η συχνότητα των φορέων στη χώρα μας φτάνει το 5%, δηλαδή κάθε 2.000-2.500 γεννήσεις έχουμε ένα παιδί που νοσεί (30 – 40 παιδιά το χρόνο). Η πιθανότητα και εδώ για ένα ζευγάρι φορέων να αποκτήσει παιδί με το ίδιο πρόβλημα είναι 25%.
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Το γονίδιο της Καλύμνου
Oνομάζεται νόσος Wilson ή ηπατοφακοειδής εκφύλιση και προσβάλλει το ήπαρ και το κεντρικό νευρικό σύστημα. Αφορά τη βλάβη ενός συγκεκριμένου γονιδίου, το οποίο ρυθμίζει τη σύνθεση και τη λειτουργία της πρωτεΐνης που μεταβολίζει το χαλκό. Και εδώ η πιθανότητα να κληρονομηθεί από ένα ζευγάρι που και οι δύο γονείς είναι φορείς στο παιδί είναι 25%. Η νόσος ενδημεί στην Κάλυμνο και σε μερικές περιοχές της Κρήτης και η συχνότητα των φορέων στα νησιά αυτά φτάνει περίπου το 5-6%, ενώ στην υπόλοιπη Ελλάδα το 1-1,2%.
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
H ΠΓΔ προσφέρει τη δυνατότητα γενετικής διάγνωσης και μεταφοράς στη μήτρα μόνο των υγιών εμβρύων που προκύπτουν με εξωσωματική γονιμοποίηση (in-vitro fertilization “IVF”). Η όλη διαδικασία απαιτεί στενή συνεργασία ενός κέντρου υποβοηθούμενης αναπαραγωγής που αναλαμβάνει και παρακολουθεί την εξωσωματική γονιμοποίηση και ενός κέντρου που είναι υπεύθυνο για τη γενετική διάγνωση της νόσου στα έμβρυα. Το Εργαστήριο Ιατρικής Γενετικής εφαρμόζει ΠΓΔ για ζευγάρια που έχουν κίνδυνο να μεταδώσουν β-μεσογειακή αναιμία, μικροδρεπανο ή δρεπανοκυτταρική αναιμία και Ινοκυστική Νόσο. Στο 2005 προβλέπεται πως θα ξεκινήσει και ΠΓΔ για φυλοσύνδετα νοσήματα όπως είναι η αιμορροφιλία και η μυϊκή δυστροφία.
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Η Ινοκυστική νόσος είναι το συχνότερο κληρονομικό νόσημα στη λευκή φυλή και κληρονομείται με υπολειπόμενο σωματικό χαρακτήρα. Φαινοτυπικά χαρακτηρίζεται από δυσλειτουργία των εξωκρινών αδένων με κύριες εκδηλώσεις από το αναπνευστικό σύστημα, το πεπτικό και τους ιδρωτοποιούς αδένες. Οι κύριες αυτές εκδηλώσεις αποδίδονται σε διαταραχές
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Το σύνδρομο Εύθραυστου Χ (FXS) με συχνότητα 1:4500 αγόρια και 1:7000 κορίτσια θεωρείται η δεύτερη πιο συχνή αιτία νοητικής υστέρησης. H κλινική εικόνα περιλαμβάνει ποικίλου βαθμού νοητική υστέρηση συνοδευόμενη από προβλήματα συμπεριφοράς όπως υπερκινητικότητα και διάσπαση προσοχής καθώς και ιδιόμορφο προσωπείο με μεγάλο μέτωπο, προεξέχον πηγούνι και μεγάλα προεξέχοντα αυτιά. Σε άρρενα άτομα παρατηρείται επίσης αυξημένο μέγεθος γονάδων (μακροορχιδισμός).
Η μοριακή διαταραχή που σχετίζεται με το σύνδρομο αφορά την επέκταση της αλληλουχίας CGG στο γονίδιο FMR-1 στο μακρύ σκέλος του χρωμοσώματος Χ στη ταινία q27.3.
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Η Μεσογειακή αναιμία, ή νόσος του Κούλεϊ (Cooley), είναι ένα βαρύτατο κληρονομικό νόσημα με μεγάλη συχνότητα στη χώρα μας όπως και σε άλλους μεσογειακούς πληθυσμούς. Χαρακτηρίζεται από βαριά αναιμία που εκδηλώνεται ήδη από τη βρεφική ηλικία με σοβαρές συνέπειες στην ανάπτυξη και την υγεία του πάσχοντος. Μέχρι σήμερα δεν έχει βρεθεί αποτελεσματική θεραπεία και η αντιμετώπιση των ασθενών περιλαμβάνει συχνές μεταγγίσεις αίματος (μια ή δύο το μήνα) αποσιδήρωση και ειδική συμπτωματική αγωγή.
Οι γονείς των πασχόντων από Μεσογειακή Αναιμία είναι οπωσδήποτε και οι δύο φορείς (ετεροζυγώτες) της νόσου. Πρόκειται για απόλυτα υγιή άτομα χωρίς κανένα σύμπτωμα που έχουν όμως το ένα από τα δύο γονίδια της αιμοσφαιρίνης τους παθολογικό (μεταλλαγμένο). Στην Ελλάδα περίπου 1 στα 10 άτομα είναι ετεροζυγώτες
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Είναι η τρίτη πιο συχνή κληρονομούμενη αυτοσωμική και επικρατής νόσος του νευρομυϊκού συστήματος με συχνότητα 1:20000. Εμφανίζεται στο τέλος της πρώτης ή δεύτερης δεκαετίας και εμφανίζει αξιοσημείωτη ετερογένεια. Τα άτομα εμφανίζουν εξελικτική αδυναμία και ατροφία των μυών του προσώπου , της ωμικής ζώνης, των άνω άκρων και σε ορισμένες περιπτώσεις των κοιλιακών, των εκτεινόντων των κάτω άκρων και της πυελικής ζώνης.
Η υπεύθυνη για το νόσημα γενετική θέση έχει χαρτογραφηθεί στη θέση 4q35 και παρότι
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
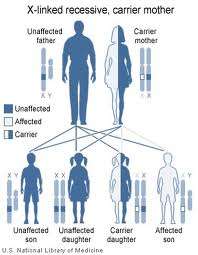
Η μυϊκή δυστροφία Duchenne είναι η πιο συχνή κληρονομική νόσος του νευρομυϊκού νοσήματος. Μεταβιβάζεται με φυλοσύνδετο υπολειπόμενο τρόπο προσβάλλοντας 1:3500 άρρενα άτομα. Η τύπου Becker μυϊκή δυστροφία οφείλεται στην ίδια γενετική ανωμαλία όπως η Duchenne αλλά κλινικά ακολουθεί ηπιότερη και πιο μακρά πορεία. Και οι δύο τύποι οφείλονται σε μεταλλάξεις του γονιδίου της δυστροφίνης (δυστροφινοπάθειες).
Στην πλειονότητα των ασθενών (65%) ανιχνεύονται ελλείψεις εξονίων του γονιδίου της δυστροφίνης που εντοπίζονται κυρίως σε δύο περιοχές του γονιδίου (hot spots) και ανιχνεύονται εύκολα
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
Ο προγεννητικός έλεγχος προσφέρει στους γονείς και τους ιατρούς τη δυνατότητα έγκαιρης ενημέρωσης για την κατάσταση υγείας του εμβρύου, οικογενειακού προγραμματισμού και αποφυγής της απόκτησης παιδιού με γενετικό νόσημα ή συγγενή ανωμαλία.
Τα κριτήρια για προγεννητικό έλεγχο είναι:
1. Η σοβαρότητα του νοσήματος
2. Η δυσκολία ριζικής ή ουσιαστικής θεραπευτικής αντιμετώπισης του νοσήματος μετά τη γέννηση
3. Η διαθεσιμότητα τεχνικής και μεθόδου αξιόπιστης, χαμηλού κόστους και χαμηλού κινδύνου τόσο για τη μητέρα όσο και για το έμβρυο
Οι ενδείξεις του προγεννητικού ελέγχου είναι:
1. Ηλικία εγκύου > 35 ετών
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
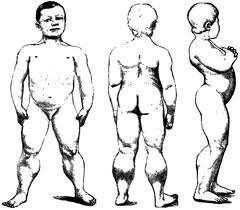
Το σύνδρομο Prader Willi (PWS) μια πολυσυστηματική διαταραχή που οφείλεται σε υποθαλαμική δυσλειτουργία αποτελεί τη συχνότερη γενετική αιτία παχυσαρκίας. Το PWS εμφανίζεται με συχνότητα 1:10-15000 γεννήσεις χαρακτηρίζεται από νεογνική υποτονία που προκαλεί διαταραχές στη σίτιση και την πρόσληψη βάρους ενώ μετά το 3ο-5ο έτος, εμφανίζεται βουλιμία και παχυσαρκία. Παράλληλα σε ασθενείς με PWS παρατηρούνται υπογοναδισμός, μικρά άκρα, χαρακτηριστικό προσωπείο με μεγάλα αμυγδαλωτά μάτια μεγάλο φίλτρο και μικρό στόμα και ψυχοκινητική καθυστέρηση με συχνά προβλήματα συμπεριφοράς. Η μοριακή διαταραχή αφορά την απουσία αλληλομόρφων πατρικής προέλευσης στη περιοχή q11-13 του χρωμοσώματος 15.
Πληροφορίες: Καθηγητής Γενετικής
Διευθυντής Εργαστηρίου Ιατρικής Γενετικής
ΠΑΝΕΠΙΣΤΗΜΙΟΥ ΑΘΗΝΩΝ
ΝΟΣΟΚΟΜΕΙΟ ΠΑΙΔΩΝ «ΑΓΙΑ ΣΟΦΙΑ»
ΧΩΡΕΜΕΙΟ
Τηλ. 210-7467468Προβολή άρθρων του συγγραφέα Γράφτηκε στις ,
(WILSON DISEASE-ATP7B)
H ηπατοφακοειδής εκφύλιση ή νόσος του Wilson (WND-ATP7B) είναι μια κληρονομική νόσος που μεταβιβάζεται με υπολειπόμενο σωματικό χαρακτήρα και οφείλεται στη μείωση της απέκκρισης χαλκού από τα ηπατικά κύτταρα καθώς και τη μειωμένη ενσωμάτωση του στη σερουλοπλασμίνη. Aυτό έχει ως συνέπεια την αύξηση των επιπέδων του χαλκού και την συσσώρευση του στον ηπατικό ιστό, και στη συνέχεια στον εγκέφαλο, νεφρό και κερατοειδή χιτώνα. Η νόσος περιγράφηκε για πρώτη φορά το 1912.
Χρησιμοποιούμε cookies για να σας προσφέρουμε την καλύτερη δυνατή εμπειρία στη σελίδα μας. Εάν συνεχίσετε να χρησιμοποιείτε τη σελίδα, θα υποθέσουμε πως είστε ικανοποιημένοι με αυτό.ΕντάξειΌροι Χρήσης